INTRODUCTION
Myotonic dystrophy (MD) is a complex genetic disorder that primarily causes progressive atrophy and muscle weakness1). MD type 1 caused by CTG repeat expansions in the 3′-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19 and MD type 2 result from CCTG repeat expansions in intron 1 of the cellular nucleic acid-binding protein (CNBP) gene on chromosome 38,11). Therefore, target therapies including antisense oligonucleotide targeting the DMPK mRNA have been tried13). People with this condition often have persistent muscle contractions and problems, such as muscle weakness, contractures, and deformities in the feet or spine, which can reduce the quality of life9). Cases where the surgical correction of spinal deformities in patients with muscle relaxation disorders did not fall under absolute contraindications have been reported, but they included a few unexpected events after surgery9,12,15). This case describes unexpected, catastrophic complications after surgery in a patient with a secondary spinal deformity caused by MD.
CASE REPORT
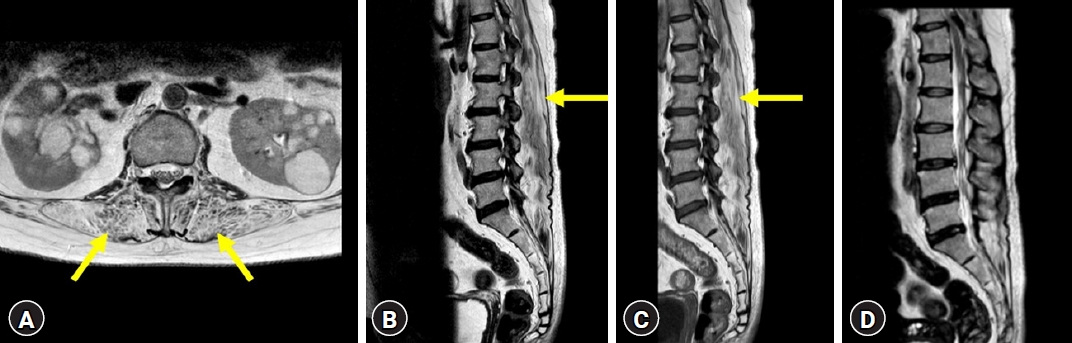
A 50-year-old female patient visited our hospital for the first time in 2017 and complained of progressive back pain, knee pain, and pain in both thighs that had lasted for more than 5 years. The initial neurological examination revealed nonspecific leg pain. In 2021, the patient’s gait length gradually decreased, and back pain worsened. In further neurologic examination, ptosis, weakness in the hand grip, a waddling gait with weakness in the hips and thighs, positive Romberg sign, impaired tandem gait, and variations in symptoms throughout the day were identified. X-ray films of the whole spine revealed less lordosis and worse positive sagittal balance. A computed tomography (CT) scan of the brain showed no abnormal findings that could explain these symptoms. Nerve conduction velocity and electromyography, as well as the Jolly test, revealed electrophysiological findings consistent with MD, including abnormal transmission at the neuromuscular junction. Magnetic resonance imaging (MRI) showed severe atrophic changes in both paraspinal muscles, manifested as decreased paraspinal muscle volume and fatty infiltration (Fig. 1). In the MRI, there were no foraminal and central canal stenosis and the disc heights at the levels of L2-3-4-5-S1 seemed to be well maintained (Fig. 1). Genetic testing confirmed the diagnosis of MD, with amplification of CTG repeats in one allele of the DMPK gene.
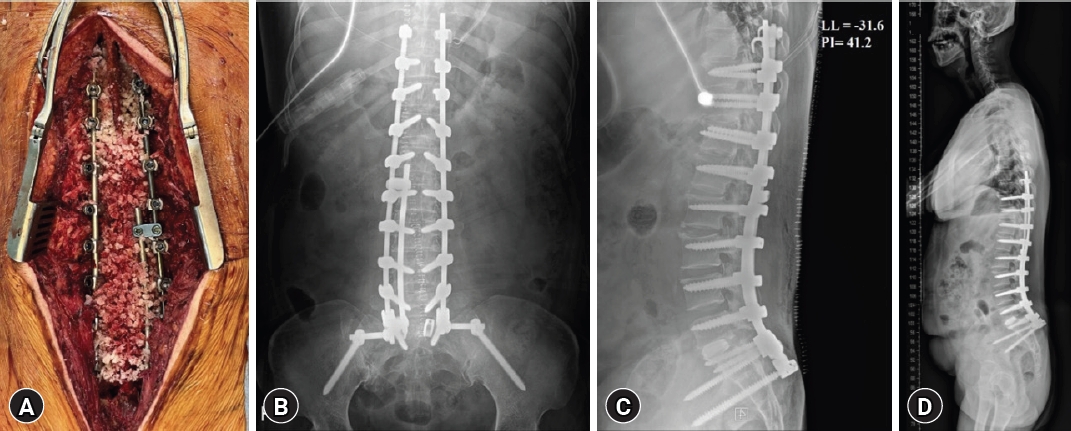
Therefore, we diagnosed the patient's paraspinal muscle weakness due to MD as the cause of kyphosis and lumbar lordotic curve seemed to be restoration in extension lumbar simple X-ray (Fig. 2). To summarize the patient’s information, the main symptoms were proximal muscle weakness, aggravative stooping posture and lower back pain without specific radiculopathy and there was no central and formal canal stenosis in imaging study. The disc heights of lumbar spine were well maintained. In addition, the patient was diagnosed with MD and had reasonable genetic and imaging test results and clinical symptoms. Therefore, we thought the spinal deformity was caused by MD. And, consecutively, we decided to proceed with surgery to correct the kyphosis, aiming to alleviate the patient's back pain. There was severe stenosis at T11-12 level due to a thickened yellow ligament, she underwent additionally T11-T12 hemilaminectomy, T12-L5 Smith-Petersen osteotomy, and L5-S1 transforaminal lumbar interbody fusion surgeries for 10 hr and 30 min (Fig. 3). No events occurred during surgery. The patient received repeated administration of phenylephrine to maintain blood pressure during general anesthesia. After intensive care unit (ICU) transfer, the continuous infusion of norepinephrine was required at a rate of 4 mcg/min due to persistent hypotension. Two hours after surgery, the patient was capable of obeying commands and had stable respiration, leading to extubating.
However, 5 min after extubation, the patient’s respiratory rate increased to 30, desaturation occurred with oxygen saturation dropping to 77%, and the norepinephrine dosage had to be increased to 32 mcg/min. Arterial blood gas analysis (ABGA) testing revealed pH 7.150-pCO2 59.7-pO2 60.5-HCO3¯ 20.3 and a SpO2 of 80%, indicating type 2 respiratory failure with primary respiratory acidosis. Consequently, immediate reintubation was performed. Before reintubation, a train-of-four test was conducted to assess muscle relaxant reversal, showing 82% reversal, which was deemed sufficient. Considering acute desaturation, hypotension, and D-dimer levels elevated to 2.70, pulmonary CT angiography was conducted to rule out pulmonary thromboembolism (PTE). The scan revealed a small PTE, which was not considered to induce significant blood pressure decline. However, patchy consolidation and passive atelectasis with a volume decrease were seen in both posterior lungs. Also, the hemoglobin blood level decreased from 13.2 to 9.0 g/dL postoperatively, and the estimated blood loss during surgery was 1,050 mL. However, the intraoperative fluid balance was +3,085 mL, and body weight increased from 64.7 to 67 kg after surgery, so volume depletion was ruled out. Abdominal aorta CT angiography was performed to exclude internal organ damage caused by the screws, and there were no abnormal postoperative findings. In summary, the patient's respiratory failure and hypotension were attributed to passive atelectasis resulting from respiratory muscle weakness and insufficient ventilation after anesthesia. Subsequently, the patient remained in the ICU, waiting for an improvement in respiratory muscle weakness. It took 5 days before the patient could undergo extubation, and even after extubation, an additional week of high-flow nasal cannula oxygen therapy was necessary to achieve sufficient tidal volume recovery. Fortunately, the demand for vasopressor agents gradually decreased, and after 3 days, she was completely tapered off these agents. The patient’s condition gradually stabilized over a 1-month period in the ICU, her oxygen support was successfully discontinued, and she was transferred to the Department of Rehabilitation Medicine for continued rehabilitation treatment. The patient showed significant improvements in strength, mobility, and respiratory function with regular rehabilitation and multidisciplinary support. The patient achieved a level of functional independence 6 months postoperatively that allowed her to perform her daily activities with minimal assistance.
DISCUSSION
The MD comprise type 1 and 24). The two forms of the disease are genetically distinct. MD type 1 is caused by an expanded CTG triplet in MPK on chromosome 19 while MD type 2 is caused by the expansion of a CCTG tetramer in CNBP2). The main symptoms of MD type 1 are ptosis, temporal wasting, weakness of facial and oropharyngeal muscles, neck flexion, respiratory muscles, truncal muscles and distal limb muscles and grip myotonia. The weakness and myotonia of proximal muscles weakness and muscle pain are the main symptoms of the MD type 24). Although we tried to find the family history for the MD, he mother of patient denied the family history related to the MD. The understanding of the underlying molecular mechanism of MD and treatments targeting the genetic cause of MD have progressed and clinical trials have been tested4). However, because the phenotype is variable and diverse, the challenge of treatment continues in clinic and in the research. Because of these symptoms of muscle weakness, the patients with MD are sensitive to general anesthesia and neuromuscular blocking agents. Therefore, although there were no abnormal findings in the routine preoperative test, clinical issues including our case associated with delayed emergence from anesthesia and postoperative cardiovascular and respiratory complication have been reported5,14). The previous study reported that general anesthesia using remimazolam and remifentanil in combination with regional anesthesia may be a good option for the patients to reduce the complication6).
Hypotonia and muscle weakness of the trunk may cause difficulty in upright positioning and lead to spinal deformities with thoracolumbar kyphoscoliosis3). Spinal deformities in patients with MD tend to develop slowly, and surgical correction can improve ambulatory ability9). Although surgical correction may not be a contraindication for patients with MD, these patients have a high risk of experiencing anesthetic complications7,10,12). The incidence of postoperative pulmonary complications was reported to be 8.2%7). Therefore, the stage and impact of the disease must be evaluated in depth, especially with respect to preoperative cardiovascular and respiratory health. Cardiac function should be assessed by routine electrocardiogram (ECG) and cardiac examinations, and there should be a low threshold for performing an ECG to assess cardiac function. Respiratory function is evaluated with additional tests such as ABGA, pulmonary function test, chest X-ray, and chest CT, considering the individual's level of muscle strength, history of dyspnea, history of anesthesia, the use of respiratory support devices, and additional risk factors.
In the present case, an ECG was performed to check for heart-related complications of MD before surgery and normal results were identified (ejection fraction = 66.1% biplane), confirming that there were no special problems in cardiac function. And despite the presence of diffuse inhomogeneous attenuation on the preoperative chest CT, surgery was considered appropriate as the patient had no active lesions or respiratory infection symptoms or signs and no special risk factors before surgery. However, postoperative hypoxemia and hypotensive symptoms were observed. The patient's cardiac markers, including creatine kinase (CK), CK-myocardial band, and troponin I, increased after surgery. However, a repeat ECG conducted postoperatively did not reveal any new ST-T changes compared to the initial ECG taken before the surgery. The pattern of cardiac enzymes was closer to a stress or type 2 myocardial infarction (MI) pattern than the typical type 1 MI. Liver function tests, including aspartate aminotransferase and alanine aminotransferase, also showed elevated levels. Given the patient's history of hypoxemia and shock, the possibility of ischemic hepatitis was considered the most probable. Cardiology, respiratory medicine, and neurology were consulted to evaluate and manage the occurrence of this situation. The patient was thought to have developed passive atelectasis after surgery due to the influence of the sedative and muscle relaxant used for anesthesia. The patient received supportive care in response to these findings to maintain blood pressure and oxygen saturation levels. The elevated cardiac and liver enzymes continued to rise until peaking 3 days after surgery, after which the patient gradually showed signs of improvement. After checking the medications administered to the patient, drugs that could affect consciousness, such as dimenhydrinate and alprazolam, were discontinued, and the patient stabilized over time with symptomatic treatment.
CONCLUSION
MD can lead to spinal deformity and cause catastrophic postoperative complications. Therefore, even if there are no special medical problems before surgery, complications and rapid deterioration of the condition may occur postoperatively. Thus, surgeons should consider the possibility of postoperative pulmonary and cardiac complications and prepare multidisciplinary support and ICU treatment after surgery on these patients.