INTRODUCTION
Rosai-Dorfman disease (RDD), also referred to as sinus histiocytosis with massive lymphadenopathy, is a rare manifestation of non-Langerhans cell histiocytosis12). RDD was first described by Destombes in 19654), based on observations of four patients exhibiting lymphadenopathy and sinus histiocytosis. While RDD is commonly associated with self-limiting bilateral cervical lymphadenopathy, approximately 43% of RDD cases display extranodal involvement6). The disease often affects the skin, eyes, head, neck, and bones, whereas its presence within the central nervous system (CNS), gastrointestinal tract, and genitourinary system is less frequent1). Notably, CNS involvement occurs in <5% of extranodal RDD cases (CNS-RDD), with spinal lesions accounting for a quarter of such instances13). In this report, we present two cases of RDD involving the thoracic and cervical spinal cord. Initial diagnosis of this rare condition posed challenges. Furthermore, we reviewed potential mechanisms contributing to spinal cord involvement.
CASE REPORT
1. First Case
A 63-year-old man was admitted to the emergency department primarily owing to an abrupt deterioration in his gait disturbance, leg weakness, and lower back pain, coupled with hypoesthesia of his legs that had persisted for two days. Similar symptoms had manifested two months earlier, leading to a one-month hospitalization at another healthcare facility. During that admission, a magnetic resonance imaging (MRI) scan revealed dural thickening with minor hemorrhagic changes suggestive of myelitis. Intravenous steroid therapy and conservative care led to symptom resolution, enabling the patient to regain independent mobility. However, a month post-discharge, the patient encountered a recurrence of primary complaints, prompting his visit to the emergency room. Gradually worsening gait disturbance culminated in an inability to walk, necessitating wheelchair assistance.
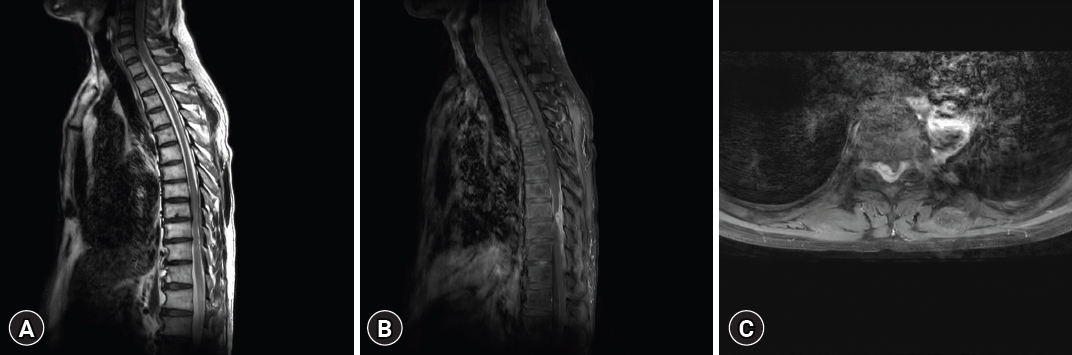
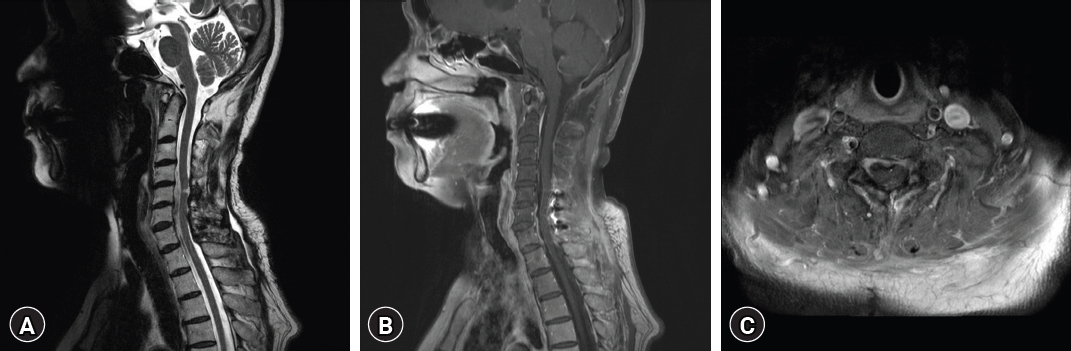
A computed tomography (CT) scan exhibited an iso-dense peridural lesion with an ill-defined margin, filling the spinal canal from T8 to T10. An MRI scan of the thoracic spine confirmed the existence of an intradural and extradural gadolinium-enhancing mass between the T8 and T10 levels, displaying a feature of cord compression (Fig. 1). Initial impressions based on the thoracic MRI included hypertrophic pachymeningitis, meningioma, spinal lymphoma, solitary fibrous tumor, or metastasis with compressive myelopathy.
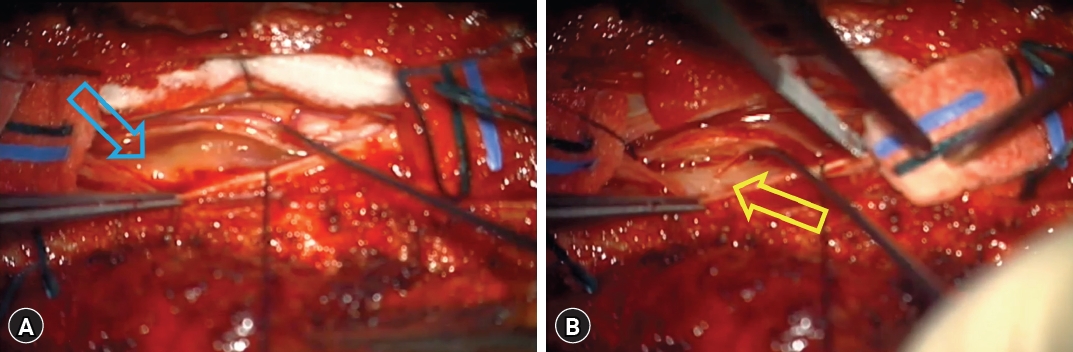
The patient underwent surgical intervention for thoracic spinal cord decompression. Post-laminectomy, an epidural mass compressing the spinal cord with adhesions was observed. Meticulous dissection of the epidural component was followed by durotomy. The intradural mass directly compressing the spinal cord was carefully detached from the dura mater using a dissector and completely excised (Fig. 2).
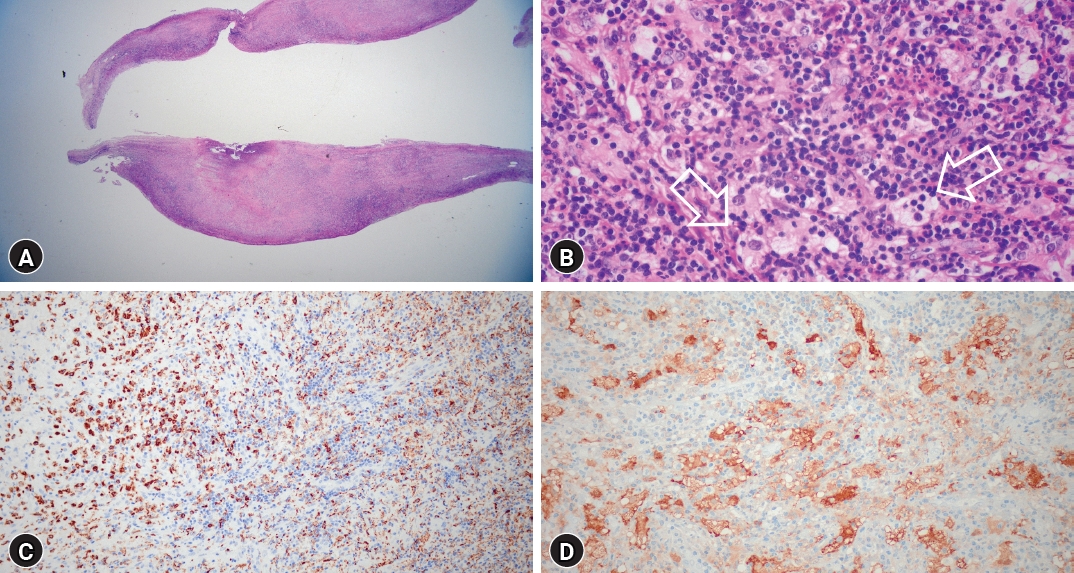
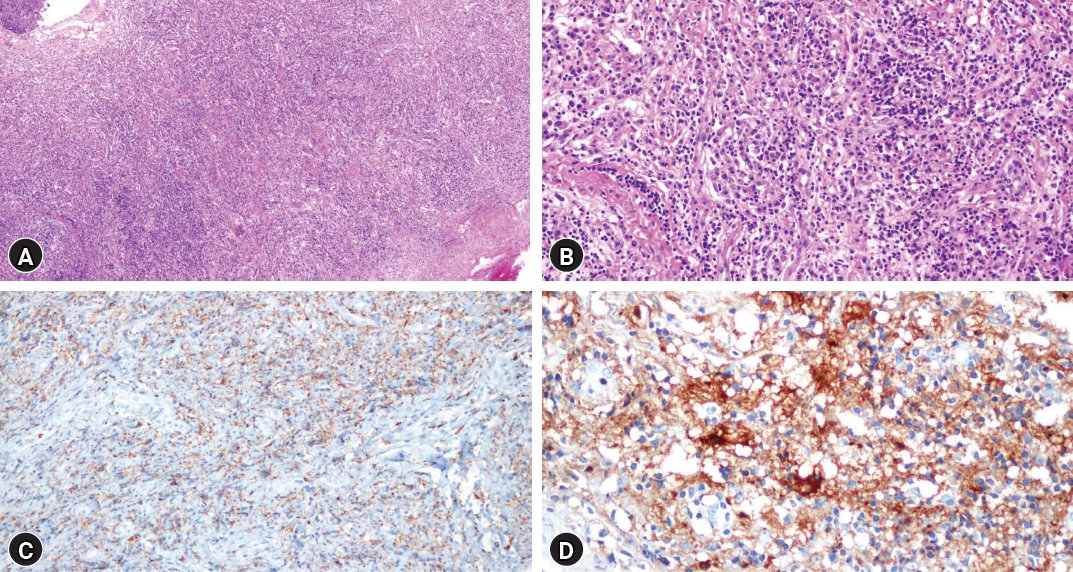
Postoperatively, bilateral leg motor function exhibited significant improvement. Bilateral leg pain subsided, allowing the patient to walk with a cane. Histopathologic examination revealed multifocal lymphohistiocytic infiltration, indicative of RDD with characteristic immunochemical staining (Fig. 3). Post-surgery, oral prednisolone therapy commenced at a daily dose of 60 mg, gradually tapered over months. A positron emission tomography (PET) scan performed one month postoperatively revealed no signs of recurrence and negative findings for systemic involvement. Similarly, a whole-body CT scan at three months postoperatively yielded identical results (Fig. 4).
2. Second Case
A 64-year-old woman presented with left shoulder and arm pain, accompanied by left-side weakness graded at 4, which had persisted for one year. MRI revealed a cervical spinal cord tumor. Notably, seven years prior to the surgery, the patient experienced mild headaches and blurred vision, subsequently diagnosed with multiple intracranial masses suspected to be meningiomas. The patient underwent her first γ knife surgery (GKS) in 2005. Two years later, progressive hearing difficulties emerged owing to a growing mass in the left intra-auditory meatus despite the stable status of the pretreated lesion. This necessitated a second GKS. Both interventions yielded substantial rapid volume responses; however, pathologic confirmation of these lesions was not secured.
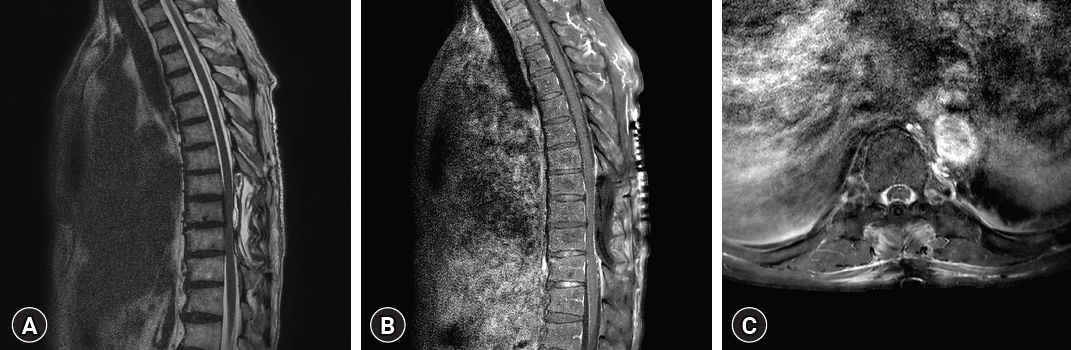
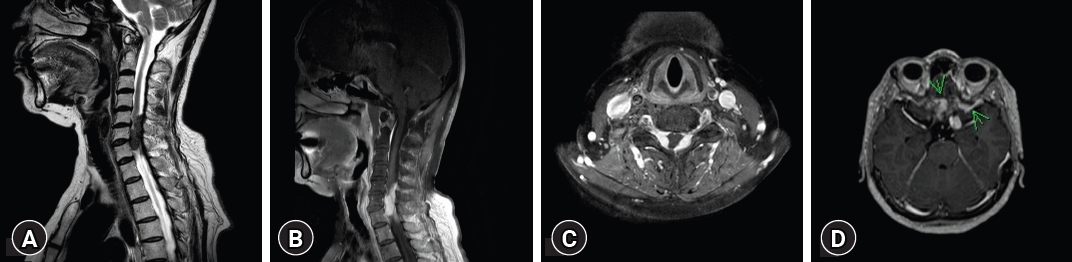
Five years following the second GKS, MRI of the cervical spine revealed two distinct dura-based intradural masses between C5 to C7, encapsulating the cervical cord. Furthermore, a separate mass displaying a characteristic dumbbell shape was noted in the left C7 root (Fig. 5). Multiple nodules were also evident in the lumbosacral region, suggesting a neurogenic tumor. Considering the coexistence of these masses in the brain, the preliminary diagnostic impression inclined towards multiple meningiomas and schwannomas, indicative of a potential neurofibromatosis.
Surgical intervention was conducted through a posterior approach. The operation included a laminectomy and durotomy, both cautiously executed. An intradural mass at the C4 level was observed bilaterally, attached to the posterior dura, and causing spinal cord compression with an en-plaque type extension. Another mass in the C7 root space had extended anteriorly, resulting in spinal cord compression and invasion of the left lateral dura wall. Alongside these two major tumors, multiple micro-tumors with dura invasion were also noted. After thorough dissection, the principal mass was completely excised. Immediately post-surgery, the patient regained leg and arm motor functions.
Histopathologic findings aligned with diffuse plasma cell infiltration, suggestive of extranodal RDD, with negative findings for IgG4-related sclerotic diseases and meningioma (Fig. 6). Postoperative MRI revealed no evidence of residual tumor in the cervical spinal cord. Following discharge, oral prednisolone therapy was initiated (Fig. 7).
DISCUSSION
RDD is a rare, benign, self-limiting condition with a prevalence of approximately 1:200,000 people and an average onset age of 20.6 years1). Recently, RDD has been added to the 5th edition of the World Health Organization classification as part of the histiocytic/dendritic neoplasms12). Neoplasms in this group are derived from common myeloid progenitors lineages12). Notably, these conditions often share mutations in MAPK pathway-related genes, such as BRAF, ARAF, MAP2K1, and KRAS8). Despite the elusive pathophysiology of RDD, molecular studies have demonstrated its polyclonal, reactive, and non-neoplastic features16).
RDD diagnosis requires a pathologic examination, characterized by large histiocytic cells displaying hypochromatic nuclei, prominent nucleoli, and "watery-clear" cytoplasm, often with abundant plentiful emperipolesis17). Immunostaining is crucial for RDD diagnosis, highlighting residual RDD in a lymphoplasmacytic background and fibrosis. RDD exhibits a unique immunophenotype, including S100, fascin, CD68, CD14, and CD163 macrophages, without CD1a or CD207 staining2). Confirming RDD histology is required for diagnosis, and appropriate clinical and radiologic context must be used to exclude primary malignant disorders that can mimic RDD histology1). RDD-associated symptoms involving the CNS arise due to mass effect, evolving over weeks to months1). In cases of spinal RDD, progressive onset of limb paraparesis, sensory deficits, gait disturbance, and urinary and bowel incontinence are common9). However, only 17% of CNS RDD cases exhibit massive lymph nodes9).
Imaging findings for RDD in the CNS are non-specific, often leading to misdiagnosis as infection, extradural hematoma, meningioma, diffuse pachymeningitis, or other metastatic tumors. RDD typically presents as a solitary extra-axial or dura-based, homogeneously enhancing, well-circumscribed mass with iso-intense to central neural structures in T1-weighted imaging (WI) and hypo- or iso-intense signals in T2-WI. Reports of intracranial RDD often highlight heterogenicity with low-intensity T2-WI22), supporting the notion of macrophage-produced free radicals during their lifecycle7). The MRI findings in our cases adhered to typical features, such as enhancing patterns and dura-based growth. However, shared characteristics within previously mentioned differential diagnoses posed challenges in suspecting RDD owing to its low incidence. Diagnosis and staging of RDD are crucial for planning treatment strategies. Fluorodeoxyglucose (FDG)-PET scans can effectively evaluate disease extent due to the FDG avidity of RDD14).
Treatment approaches for CNS RDD are individualized lacking a unified strategy1). While spontaneous remission occurs in 20% to 50% of nodal or cutaneous RDD cases18), self-limitation within CNS-involved RDD instances is exceedingly rare. Surgical resection is often the preferred treatment choice, particularly for unifocal disease confined to curative options, owing to common symptoms associated with CNS RDD11). When the neoplasm is unresectable, debulking for symptom control can be beneficial11). Considering the potential for recurrence, several case studies indicate a slightly diminished recurrence rate in cases where total resection was achieved compared with those in which it was not achieved9). Corticosteroids are usually effective for size and symptom reduction. However, the ideal dosage and duration remain undetermined. A dose of 40 to 70 mg/day of prednisolone has demonstrated complete or partial responses in CNS RDD cases, which generally involves a higher treatment dosage than other conditions that require long-term corticosteroid therapy5,21). Other therapeutic options, including chemotherapy, immunomodulatory therapy, targeted antibodies, and radiotherapy, targeting gain-of-function mutations in the MAPK pathway, can be used in refractory and recurrent RDD cases15,18,19).
The cases in our report required unique presurgical treatments. The first case required intravenous corticosteroid administration, leading to symptom alleviation. The second case required GKS for brain lesions, yielding a favorable outcome. While positive symptoms and radiographic improvements were evident, the intervention commenced before an accurate diagnosis was established. Swift reduction of mass effect, which might not have been achieved, could potentially influence the disease progression and result in neurological sequelae. These instances emphasize the necessity for surgical resection or, at minimum, pathologic confirmation via biopsy, especially in spinal cord lesions presenting with neurological symptoms.
The unique aspect of these cases is their multi-location occurrence. Current molecular analyses of RDD indicate a lymphatic origin for the primary reactive process13). Hence, CNS-involved RDD sheds light on the structure and anatomy of CNS lymphatics. Recent studies highlight perineural and perivascular spaces as crucial pathways for cerebrospinal fluid (CSF) outflow into extracranial lymphatics20). Investigating the role of cranial nerve sheaths in these channels is ongoing10). Although CSF clearance through lymphatic vessels accumulating into the sacral and iliac lymph nodes in the lower spine has been demonstrated, comprehensive anatomical understanding remains limited3). The first case suggests dura mater-based lymphatic drainage at the thoracic level. In the second case, while only the cervical cord lesion was confirmed as RDD pathologically, the intracranial lesion and multiple masses in lumbosacral lesion mimicking meningioma and neurogenic tumor might also be RDD, implying a distant propagation of histiocytic neoplasm. Considering the potential dual pathways for disease spread—through the lymphatic system and CSF space—elucidating the precise dissemination mechanisms could offer viable therapeutic targets for neoplasms spread through CNS space. These cases provide valuable insights into potential lymphatic pathways and the possibility of distant transmission of histiocytic neoplasms, further emphasizing the importance of understanding the dissemination mechanisms.
CONCLUSION
In conclusion, RDD with CNS involvement is rare and poorly understood. These two unique cases highlight the complex nature of this disease, which underscores the challenges in diagnosis and treatment, particularly due to its resemblance to other conditions. Surgical intervention for complete disease eradication, followed by corticosteroid therapy, has yielded promising outcomes. These instances also raise the prospect of RDD serving as a marker for comprehending the lymphatic anatomy and dynamics of the CNS, thereby presenting novel therapeutic opportunities for other neoplasms with similar disseminating patterns. Future research to explore the characteristics of RDD and its potential role in elucidating unfamiliar aspects of CNS lymphatics is warranted.