INTRODUCTION
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare embryonal tumors found in patients <3 years old15). There is no standard treatment regimen for AT/RT to date1). According to a study, 91%, 74%, and 42.4% of patients underwent surgery, chemotherapy, and radiation therapy respectively. The overall survival (OS) is 14.3 months (11.9-16.6 months), and the 5-year OS rate is 29.9%4). We present an unusual case of a patient that showed rapid regrowth at 1 month after gross total resection (GTR), and the patient died a month later.
CASE REPORT
A 22-year-old man was admitted with a severe headache, left ear fullness with hypoacusis, dysphagia, left facial palsy (House-Brackmann grade III), left sternocleidomastoid muscle weakness (Medical Research Council [MRC] grade 4), right-side deviated uvula, decreased left anterior taste and left trapezius muscle weakness (MRC grade 4).
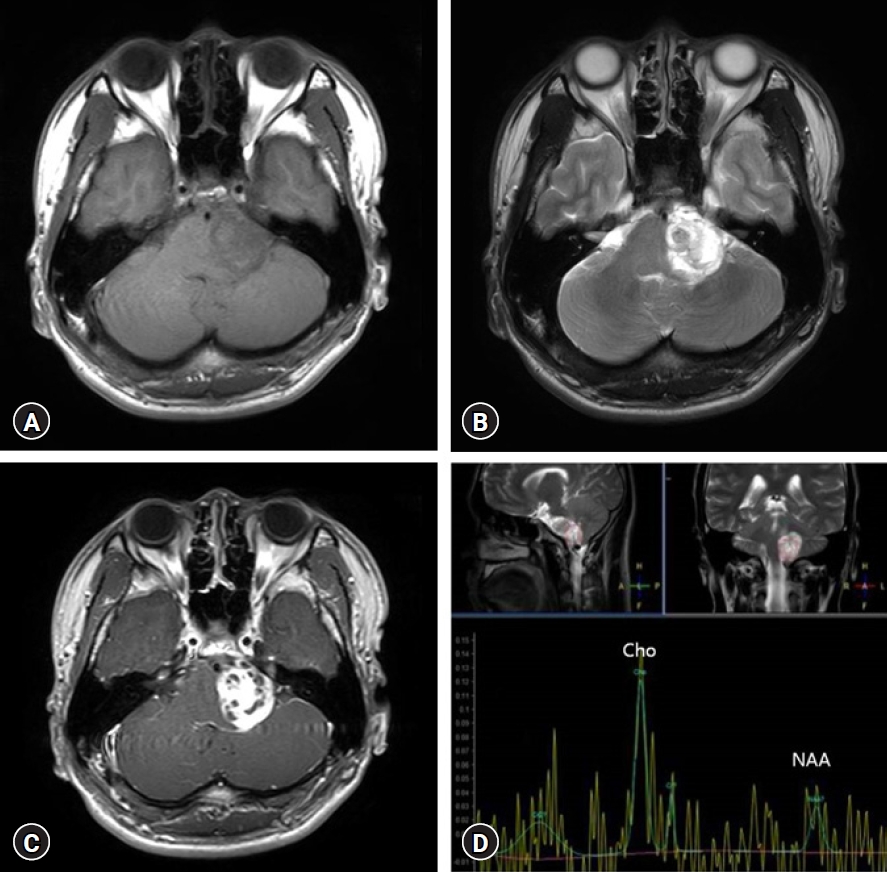
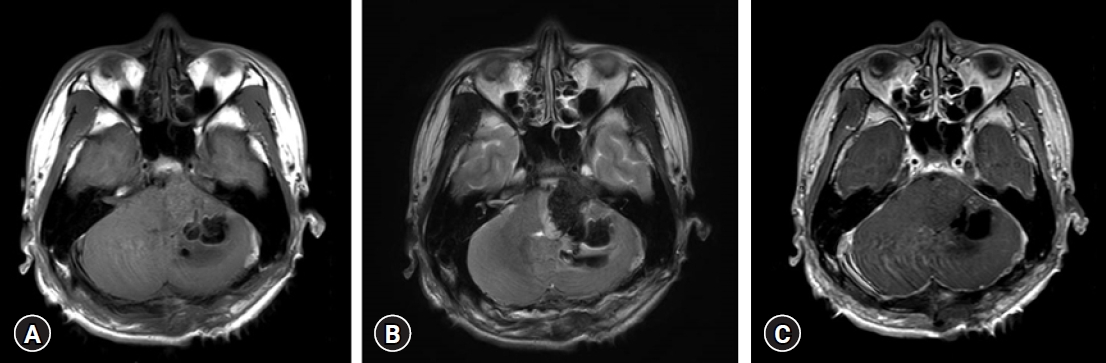
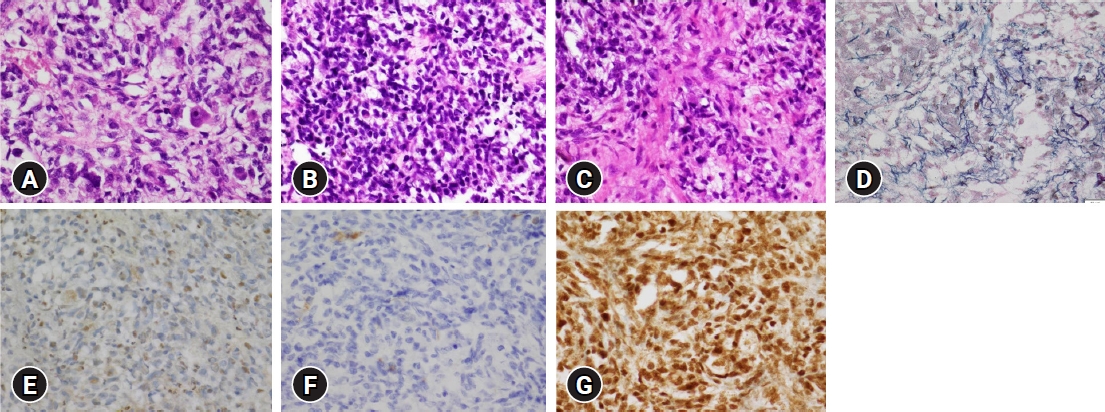
Magnetic resonance imaging (MRI) represented, a well-marginated, 2.8 × 3.7 × 3.7 cm (38.3 cm3) sized tumor in left cerebellopontine angle, which had both solid and cystic portions (Fig. 1A-C). Magnetic resonance spectroscopy showed a choline peak, decreased N‐acetyl aspartate (Cho/NAA: 2.01), and creatine, and increased lactate/lipid, suggesting malignant nature (Fig. 1D). The tumor was then removed via a retromastoid approach, but cerebellar intracranial hemorrhage occurred. So decompression by suboccipital craniectomy with navigation-assisted hematoma removal was performed and operation was successful (Fig. 2A-C). After GTR, the patient’s gag reflex mildly decreased, but the headache improved, and no additional neurologic symptoms were observed. And AT/RT was histologically confirmed (Fig. 3A-G).
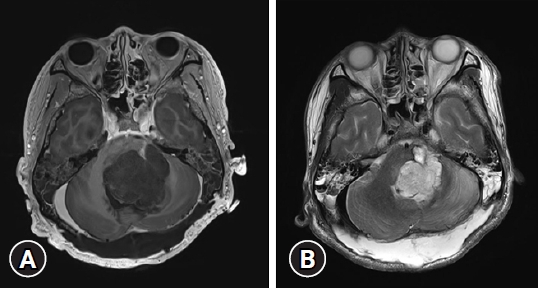
While planning the treatment regimen including chemotherapy and radiation therapy with oncologists, the patient experienced abrupt mental deterioration to comatose. On follow-up MRI, the tumor was found to have regrown to size 3.6 × 3.5 × 5.3 cm (66.78 cm3), which was even larger than its original size (Fig. 4A, B). This was only a month after the surgery. Hence chemotherapy and radiation therapy could not be considered. Subsequently, secondary tumor removal with external ventricular drainage was performed because of obstructive hydrocephalus. However, hypoxic brain injury progressed with severe brain swelling, and the patient died 1 month after the event.
DISCUSSION
AT/RTs mainly occur in children under 3 years of age and are very rare embryonal tumors, accounting for 1% to 2% of all pediatric central nervous system tumors, classified as World Health Organization grade IV11,16). They commonly occur in the posterior fossa in children; however, they manifest supratentorially in adults17). According to Ostrom et al.12), incidence rates do not significantly differ by gender, race, or ethnicity. However, age has an effect on incidence. The incidence was highest at <1 year, and it decreased with age12). The median age at the time of diagnosis was 37 months14). Patients whose ages were less than 6 months had the worst prognosis14). Moreover, it is known that the prognosis for AT/RT is better in childhood than in adulthood2).
In diagnosing AR/RTs, there are key genes in addition to biopsy and imaging studies: SMARCB1 and SMARCA410). Bi-allelic mutations in SMARCB1 (INI1) at 22q11.2 are common, whereas SMARCA4 (BRG1) mutation at 19p13.2 is rare16). The SMARCB1 gene acts as a tumor suppressor gene and inactivation of both copies of this gene causes loss of protein expression in the nucleus in immunohistochemistry (IHC)13). These genes affect the prognosis. The age of onset of SMARCA4 mutated AT/RT is lower than that of SMARCB1 mutated AT/RT, and the survival period is 3 months. However, survival period of SMARCB1 mutated AT/RT is approximately 24 months6,8).
There is no standard regimen for AT/RT treatment; therefore, radiation therapy, chemotherapy, and surgical resection have been attempted7). It usually takes approximately 50 days to start chemotherapy3). Furthermore, radiation therapy takes an average of 93 days4). Table 1 summarizes some articles on AT/RT OS.
In addition, many studies on the survival period and rate of AT/RTs related to therapeutic modality. The AT/RT postoperative median survival is 20 months, and the disease-free survival is 14 months7). Lafay-Cousin et al.9) reported event-free survival of 14 months and median survival of 20 months in 20 patients who underwent GTR9). Schrey et al.14) demonstrated that the median recurrence-free survival and OS of patients who received radiotherapy were statistically higher than that of patients who did not receive radiotherapy 12.1 (5.216-7.034) and 23 (12.39-17.61) vs. 4 (3.314-4.686) and 10 (7.803-12.197), respectively14). According to Fossey et al.5), high-dose chemotherapy exhibits better outcomes than conventional chemotherapy (5-year survival rate of 52.0% and 10.7%, respectively5).
In our case, it was an adult-onset AT/RT, and the surgery was successful. Although genetic testing was not performed, SMARCB1 mutation was suspected based on IHC results. However, within 1 month after GTR, the tumor regrew to approximately twice the original size, and the patient died before chemotherapy or radiation therapy could be initiated. In our experience, AT/RT can progress rapidly even in patients with good prognosis factors. Physicians should always consider the possibility of rapid regrowth of AT/RT.