INTRODUCTION
Hemangioblastoma (HB) is a slowly growing benign tumor of the central nervous system(CNS), belonging to World Health Organization grade I tumor. HBs usually occur in the cerebellum, brainstem, spinal cord and may occur any site of the neuraxis involving the retina5). It may occur sporadically or in association with von Hippel-Lindau (VHL) disease. Multiple HBs are typically associated with VHL disease and continue to arise over the course of the patient’s life, which necessitates life-long surveillance5). Hemangioblastomatosis is a very rare but aggressive type of the disease and has been reported in patients with or without VHL disease. The majority of published cases about hemangioblastomatosis without VHL disease were supposed to as disseminated form after surgery4). The outcomes of hemangioblastomatosis are very poor due to the absence of established treatment and the limited response to the available treatments. We report an extremely rare case which presented with hemangioblastomatosis before surgery in a patient with VHL gene mutation negative. We also add some beneficial effect of bevacizumab therapy in this particular case with literature review.
CASE REPORT
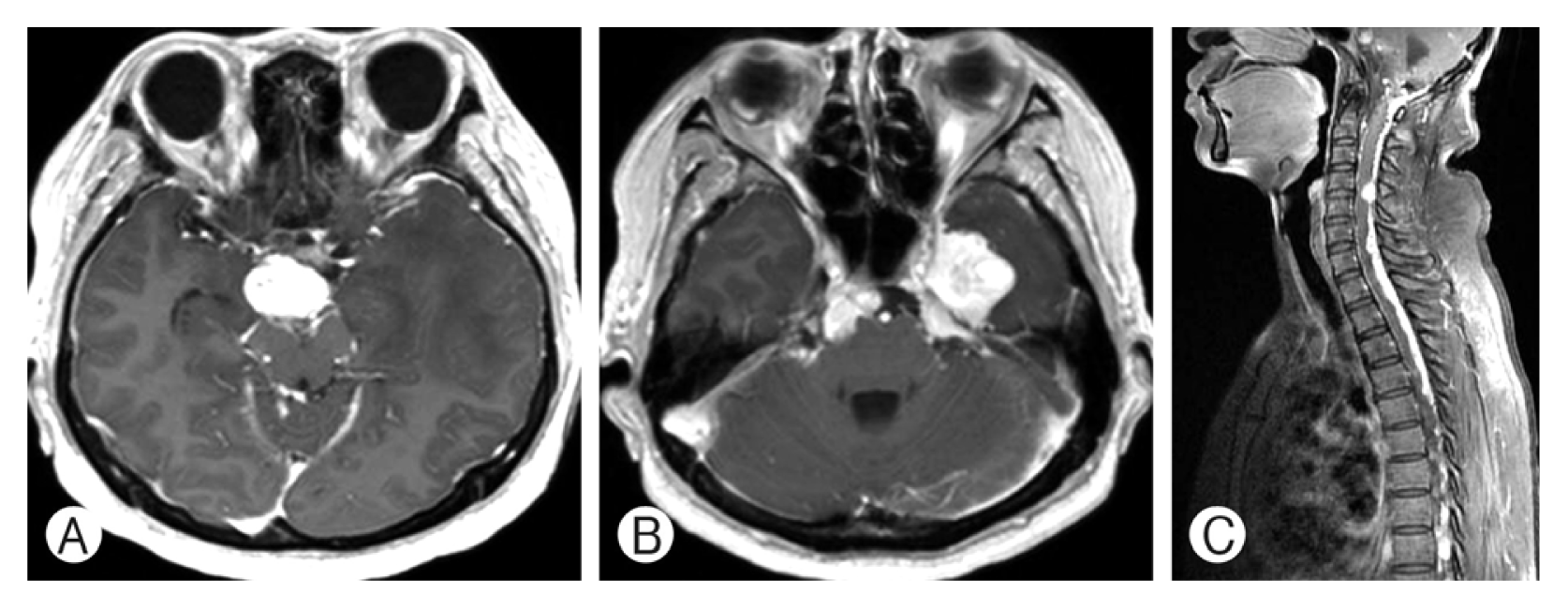
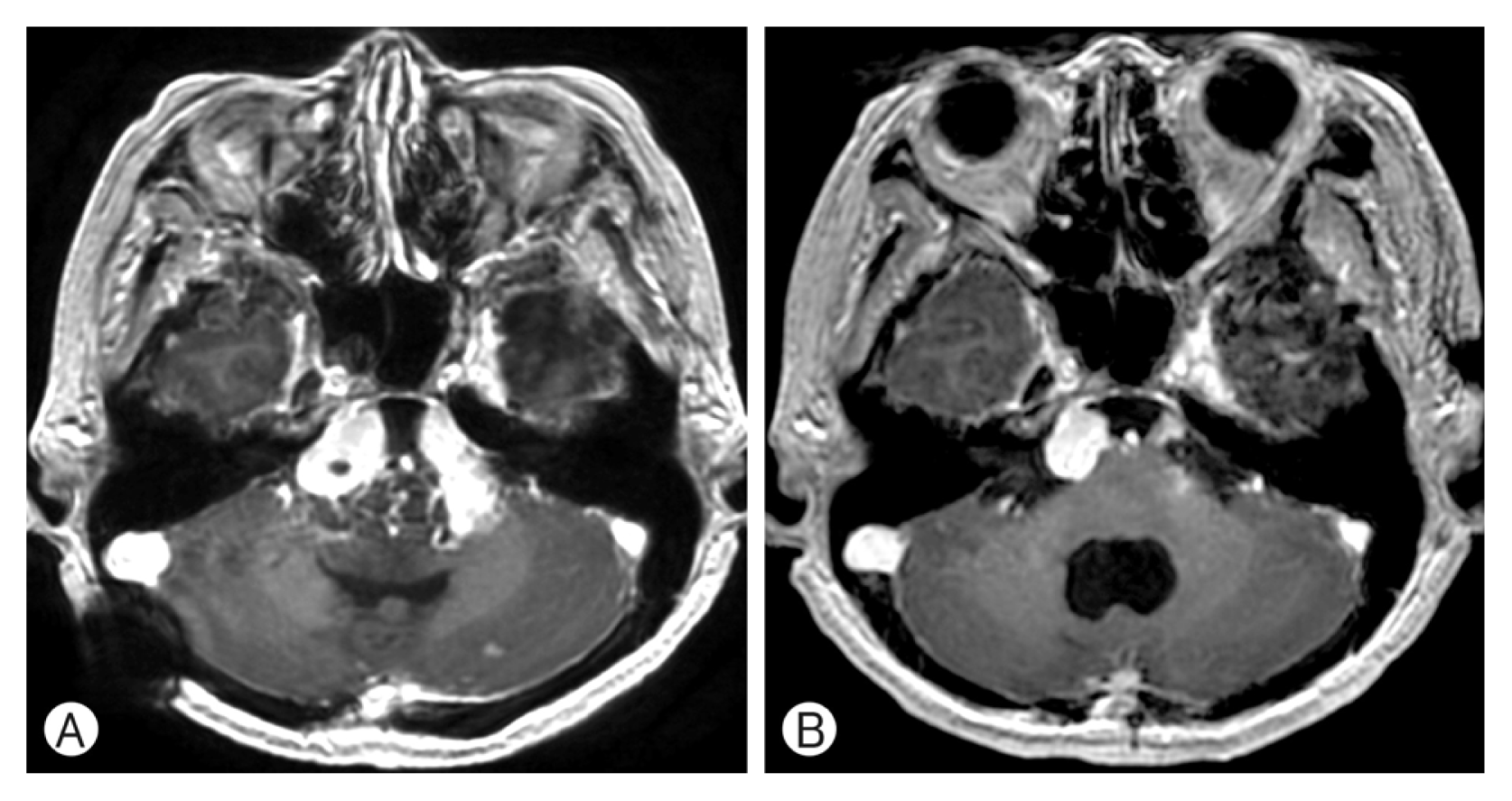
A 55-year-old woman presented with severe headache and dizziness in 2010 showing a large cerebellomedullary well enhancing tumor and a small homogenously well enhancing tumor at the left medial temporal base on MRI (Fig. 1A, B). She underwent a surgical resection for the large cerebellar mass and the pathological diagnosis was HB (Fig. 2). She had no family history of VHL disease, and the evaluation for VHL disease was negative. Because the radiological features of the left temporal lobe tumor were compatible with meningioma, we initially thought that the patient had a sporadic form of HB. Postoperative course was uneventful but one month after surgery, she complained of neck pain and was investigated with spine MRI. The spine MRI showed multiple enhancing nodules in the cervico-thoracic spine (Fig. 1C) and the patient was included in the criteria for the diagnosis of VHL disease. Although her neck pain was relieved soon after the spine MRI, we advised her regular follow-up for the left temporal tumor and the intraspinal tumors. Unfortunately, she had been lost to follow-up until December 2014, when she complained diplopia, left facial hypoesthesia and truncal ataxia. On the brain and spine MRI, multiple enhancing tumors were observed in the bilateral cerebellopontine angle, prepontine cistern exten ding to the suprasellar cistern, left parasellar area, and the leptomeninges around the whole spine (Fig. 3). She underwent surgical resection for the markedly enlarged left medial temporal tumor. The histological diagnosis was HB, which was identical to the cerebellar tumor. Her symptoms relieved after surgery. However, her cognitive function gradually deteriorated and a serial CT scans showed progression of communicating hydrocephalus. The patient underwent ventriculoperitoneal shunt (VPS) at 1 month after the second surgery.
After temporary relief of her neurological status following VPS, her condition rapidly deteriorated three months after the shunt. A follow-up MRI showed marked enlargement of the residual tumors with compression of the brainstem (Fig. 4A) and an increased extent of compressive myelopathy of the cervico-thoracic spine. Based on clinical and radiological progression, we decided to treat the patient with bevacizumab (Avastin®). Bevacizumab was administered at a dose of 10 mg per kilogram of body weight every 2 weeks. After first administration of bevacizumab, she noted improved mental status, cognitive function. Three more cycles of bevacizumab were administered and her condition improved further, and the follow-up brain MRI showed the lesions remained stable (Fig. 4B).
DISCUSSION
HB is an uncommon vascular tumor of the CNS and not infrequently associated with VHL disease, and more commonly appear as a sporadic tumor in the absence of VHL disease1-4,6,9).
VHL disease is an autosomal dominant neoplasia syndrome that results from a germline mutation in the VHL gene, tumor suppressor gene located on chromosome 3p25-p265). Diagnosis of VHL disease is often based on clinical criteria. Patients with a family history, and a CNS HB including retinal HBs, pheochromocytoma, or clear cell renal carcinoma are diagnosed as VHL disease. Those with no relevant family history must have two or more CNS HBs, or one CNS HB and a visceral tumor to meet the diagnostic criteria5). HBs in VHL disease occur in 60-80% of patients usually presenting with multiple tumors in the posterior fossa and the in the spinal cord. Supratentorial HB is a very rare and has been reported less than 1% of the VHL patients.
Multiple HBs typically associated with VHL disease are clinically benign and continue to arise over the course of the patient’s life, which necessitates life-long surveillance. However, the disseminated form of HB after surgery, which was first reported by Mohan et al. in a non-VHL patient at 1976, is a very aggressive and was named as “hemangioblastomatosis”2,6). Published cases of hemangioblastomatosis have similarly aggressive clinical and radiological presentation and mostly were not associated with VHL disease1-4). Hemangioblastomatosis is an extremely unusual disseminated form of HB, characterized by extensive leptomeningeal dissemination, causing severe neurological deficits. Most patients underwent surgical removal of the primary solitary lesion, and hemangioblastomatosis subsequently developed after variable intervals ranging from 7 months to 22 years. The outcomes after dissemination were very poor and most patients died within 3 years of diagnosis. The most common cause of death was respiratory failure due to pontomedullary or cervical cord compression1,4).
The exact mechanism of hemangioblastomatosis is unknown. There are fourteen published cases on hemangioblastomatosis and 11 cases had no relationship with VHL disease. The dissemination of HB in all reported cases occurred after surgical resection. Accordingly, the spillage and spread of tumor cells through the cerebrospinal fluid(CSF) space are the mainstreams that explain the dissemination of HB4). However, our case is different with previously reported cases. Although spinal dissemination in our case was detected at one month after surgical resection for cerebellar HB, it is reasonable to assume that the spinal dissemination is de novo lesion unrelated to surgery. Furthermore, initial brain MRI in our case showed a separate supratentorial enhancing mass that was diagnosed with HB afterward. If tumor cells dislodge from the primary site in the fourth ventricle, the majority of them will descend along the CSF flow to the spinal subarachnoid space and then ascend to the supratentorial subarachnoid space. Therefore, the initial presence of supratentorial HB suggests that our case is the de novo hemangioblastomatosis and compatible with VHL disease. Secondary, one month is insufficient time to form a multiple masses. In the literature review, the average time interval between initial operation and dissemination was 8.3 years and the shortest time interval was 7 months2,6,9).
Our patient was initially thought to have sporadic form of CNS HB, because satellite supratentorial lesion was rare in VHL disease and was supposed to as other pathologic lesion such as meningioma. A negative family history, negative other visceral and ocular lesion and a lack of manifestations by the age of 55 years suggested that the dissemination of the hemangioblastoma was not associated with VHL disease. A diagnostic challenge arises in de novo cases of VHL disease. The initial mutation in a de novo case might result in disease mosaicism. Thus, such patients might have clinical signs of the disease, but test negative genetically, because the VHL gene mutation is not carried in all peripheral leucocytes5).
The current available treatment did not have a significant effect on the progression of the hemangioblastomatosis in reported cases. Conventional radiotherapy or stereotactic radiosurgery was often used for disseminated lesions, however, long-term tumor control was not achieved and it is difficult to perform high-dose radiation therapy or radiosurgery for numerous lesions scattered throughout the entire neuraxis3,4,7-9). Some studies reported notable achievements in the treatment of hemangioblastomatosis using growth factors antagonist or blocker, such as sunitinib or erlotinib, but long term disease control was not achieved7,8). We used several cycles of bevacizumab and the patient’s neurological condition improved significantly. Long term follow-up was not established and further investigation is needed.
CONCLUSION
We report a rare case that initially presented with hemangioblastomatosis in a patient without VHL gene mutation. After four years of silent period this patient presented again as very aggressive hemangioblastomatosis. After literature review we treated her with bevacizumab resulting in acceptable clinical and radiologic outcomes.