INTRODUCTION
Hypophysitis is a relatively rare disorder caused by inflammatory infiltration of the pituitary gland that can cause tissue destruction, transient or permanent gland dysfunction, and endocrinopathies3-5). Hypophysitis has 4 histopathological subtypes: lymphocytic, granulomatous, xanthomatous, and necrotizing16). Among these, granulomatous hypophysitis is the second most common subtype and features discrete granulomas of multinucleated giant cells and histiocytes with surrounding lymphocytes and plasma cells8,25). Granulomatous hypophysitis can occur as a primary phenomenon or secondary to systemic disease. Secondary causes of hypophysitis include systemic disease (e.g., sarcoidosis, Langerhans cell histiocytosis, Wegener’s granulomatosis, disseminated tuberculosis, and syphilis), an underlying pituitary lesion (e.g., Rathke cleft cyst and adenoma), and certain medications (e.g., ipilimumab)16,27). A diagnosis of idiopathic granulomatous hypophysitis (IGH) is made when no other inflammatory cause can be identified. Here, we report a case of histologically confirmed IGH involving sellar mass lesions and pituitary dysfunction.
CASE REPORT
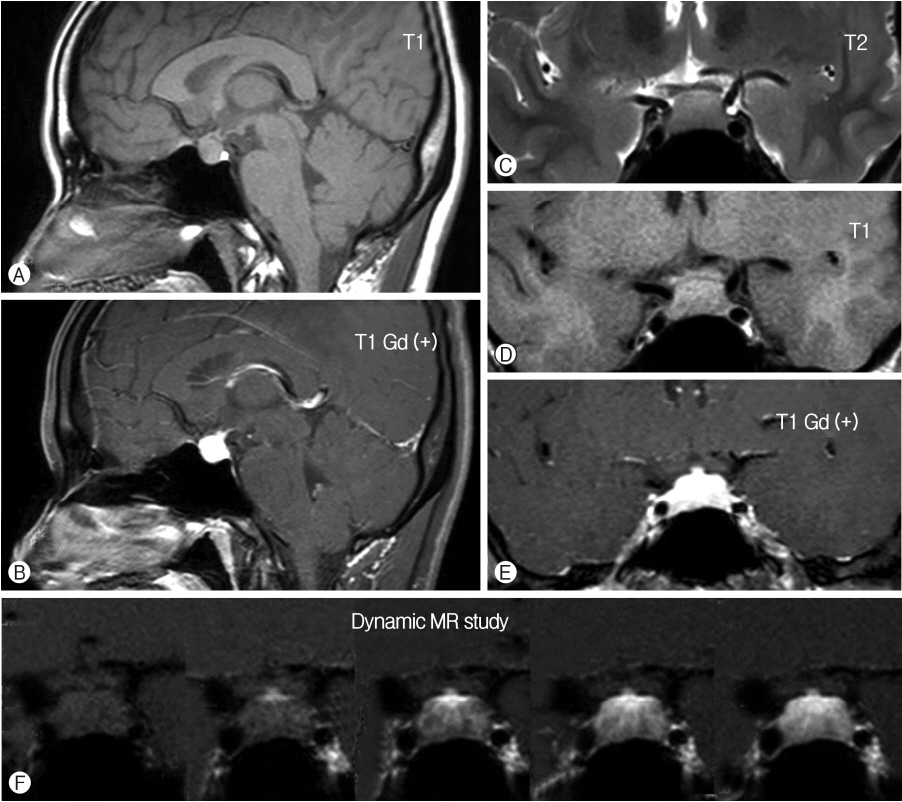
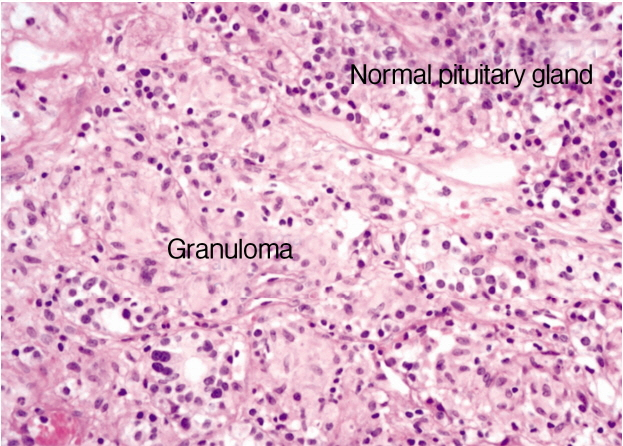
A 24-year-old woman presented with complaints of blurred vision and headache. On physical examination, the visual fields, fundoscopy findings, and neurological examination results were unremarkable. Endocrinological evaluation revealed hyperprolactinemia of 34.3 ng/mL (normal, 5.3-22.2) and otherwise nonspecific findings. Magnetic resonance imaging (MRI) showed a symmetric sellar mass that was 17 mm in size and produced mild upward displacement of the optic chiasm. The mass was isointense with gray matter on T1-weighted images (WI) and T2-WI and exhibited homogeneous gadolinium enhancement (Fig. 1). Accordingly, the patient was initially diagnosed with pituitary adenoma and secondary hyperprolactinemia. The patient underwent surgical exploration via a transsphenoidal approach. Intraoperative examination revealed that the whole pituitary fossa was occupied by a fibrous yellowish mass that adhered to the adjacent dural mater. It didn’t have a clear cleavage between inflammation and the surrounding pituitary gland. Therefore, only a partial resection was achieved under neuronavigation guidance. Histological examination revealed a non-necrotizing granulomatous lesion with chronic inflammation (Fig. 2). After histological diagnosis of the lesion as granulomatous hypophysitis, the patient further underwent cutaneous, skeletal, visceral, and laboratory examinations to exclude systemic granulomatous disease. Additional examinations included interferon-γ release assays (IGRA) for tuberculosis, antineutrophil cytoplasmic antibody and anti-nuclear antibody for vasculitis, angiotensin converting enzyme (ACE) and chest computed tomography (CT) for sarcoidosis. Given the absence of systemic disease, the patient was finally diagnosed with IGH.
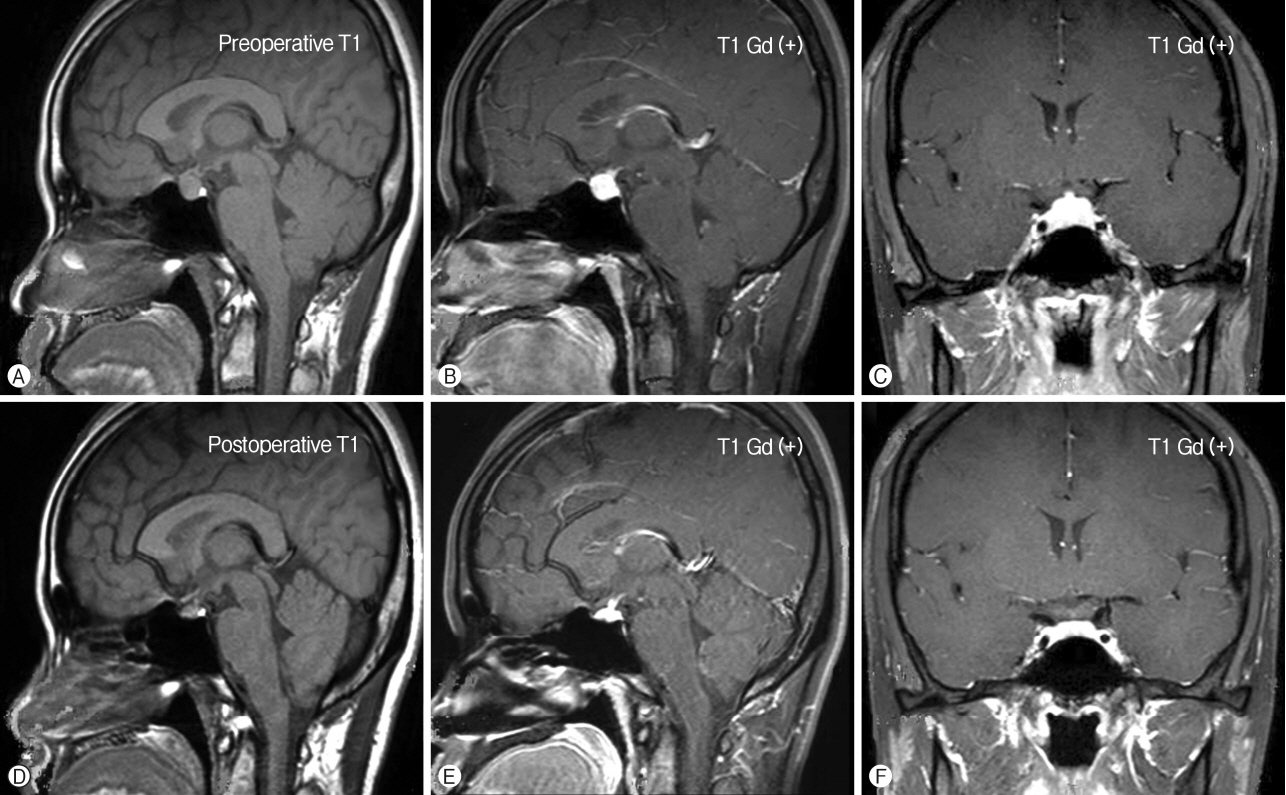
The postoperative course was uneventful. The patient’s symptoms gradually resolved and steroid treatment was gradually stopped. Although the surgical resection was limited, MRI at 6 months follow-up revealed a marked reduction in size of the sellar mass and dramatic resolution of the pituitary lesion (Fig. 3).
DISCUSSION
IGH is a rare chronic inflammatory disorder of the pituitary and accounts for less than 1% of all pituitary disorders22). Although it is generally thought that IGH has no sex predilection2,19), Su et al.24) reviewed 38 cases of IGH and reported female predilection with an approximate ratio of 2:1 (26:12). A recent systematic review of 82 cases found that 72% of patients were women14). In these studies, the mean ages of presentation were 46.1 years and 44.4 years, respectively14,24). The clinical presentation of IGH is usually that of an expanding pituitary mass lesion with insidious onset and a progressive course. The most common presenting symptom of IGH in the literature was headache, followed by visual disturbances and symptoms related to diabetes insipidus. Other symptoms included vomiting, nausea, extraocular muscle paralysis, fever, and galactorrhea. Given an insidious clinical course mimicking pituitary adenoma and acute progress mimicking acute meningitis1,7,26,29), it is difficult to correctly diagnose IGH on admission.
IGH typically presents with diffuse enlargement of the pituitary mass and enlargement of the pituitary stalk. At present, MRI is the most useful tool for diagnosing IGH and has distinct advantages over CT for imaging pituitary and sellar regional lesions. Based on a review by Su et al.24), MRI findings of primary hypophysitis include: (1) diffuse, ill-defined, symmetrical enlargement of the pituitary tissue; (2) thickening of the pituitary stalk without deviation or impaired visualization; (3) an intact sellar floor; (4) isointensity with the surrounding gray matter on T1-WI and marked homogeneous or heterogeneous enhancement by gadolinium with a strip of enhanced tissue along the dura mater (the so-called “dural tail”); and (5) delayed complete contrast enhancement of the whole pituitary on dynamic MRI (>90 s)1,6,11,13,18,20,21,24,28). Among these MRI findings, the best predictors of primary hypophysitis are diffuse pituitary enlargement, marked enhancement of the pituitary mass, and pituitary stalk thickening24).
A diagnosis of IGH should exclude secondary granulomatous hypophysitis. The etiology of secondary granulomatous hypophysitis includes infection (e.g., tuberculosis, syphilis, and fungal infection), systemic inflammatory conditions (e.g., sarcoidosis, Wegener’s granulomatosis, Takayasu’s arteritis, Crohn’s disease, and histiocytosis X), and foreign body reactions (e.g., ruptured Rathke’s cyst and mucocele)15). Despite diagnostic advances, the differentiation of primary and secondary cases of granulomatous hypophysitis remains dificult. In cases where granulomatous hypophysitis is suspected, all available steps should be taken to exclude possible etiologies of infection or systemic disease, including Mantoux testing, chest radiography, tuberculosis PCR of cerebrospinal fluid and pituitary tissue, serum syphilis testing, and serum ACE measurement.
The possible etiology of IGH is not well understood and the first-line treatment is controversial. Some studies have reported satisfactory responses to high-dose steroid therapy14,15,17). Conservative management with close clinical observation has also been advocated as a therapeutic option given the often benign and transient course of the disease and occasional spontaneous remission9). Yet, transsphenoidal surgery has both diagnostic and therapeutic utility, and should be performed in cases of progressive compression, and especially in cases of atypical clinical or radiological presentation or an unconfirmed diagnosis. Additionally, a majority of reported IGH cases and approximately half of primary hypophysitis cases were preoperatively misdiagnosed as pituitary adenomas in previous reports, similar to our case10,17,23). Surgery provides tissue for histological diagnosis and allows rapid decompression of the mass lesion, thereby resolving headache and visual defects12,13). Furthermore, surgery and definitive histological diagnosis can prevent the unnecessary use of high-dose steroid therapy and facilitate the appropriate treatment of other conditions such as infection or neoplasm. Nonetheless, one study reported no significant difference in the outcomes of patients who underwent surgical excision versus those who underwent pituitary biopsy and corticosteroid treatment14). These are all important issues for consideration when diagnosing and treating patients with IGH.
CONCLUSION
IGH is a rare inflammatory disease of unknown etiology that is easily misdiagnosed as pituitary adenoma or pituitary infection. The outcome of surgery for IGH is favorable, providing a histological diagnosis and immediate mass reduction, but hormone replacement is frequently required and long-term follow-up is very important.