INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal-dominant hereditary disease with a prevalence of about 1 in 3000. The clinical characteristics of NF1 are café-au-lait spots, neurofibromas, freckling, optic gliomas, and Lisch nodules4,5). Neurofibroma is a benign peripheral nerve sheath tumor (PNST) that can transform into atypical neurofibroma, a premalignant tumor, or malignant PNST (MPNST)1,9). Because NF1 patients are at higher risk of malignancy, which is the most common cause of death, surgical resection and pathologic confirmation are very important to the treatment of symptomatic or growing tumors in NF1 patients3,4,11). We present three cases of different PNSTs in NF1 patients.
CASE REPORT
Case 1
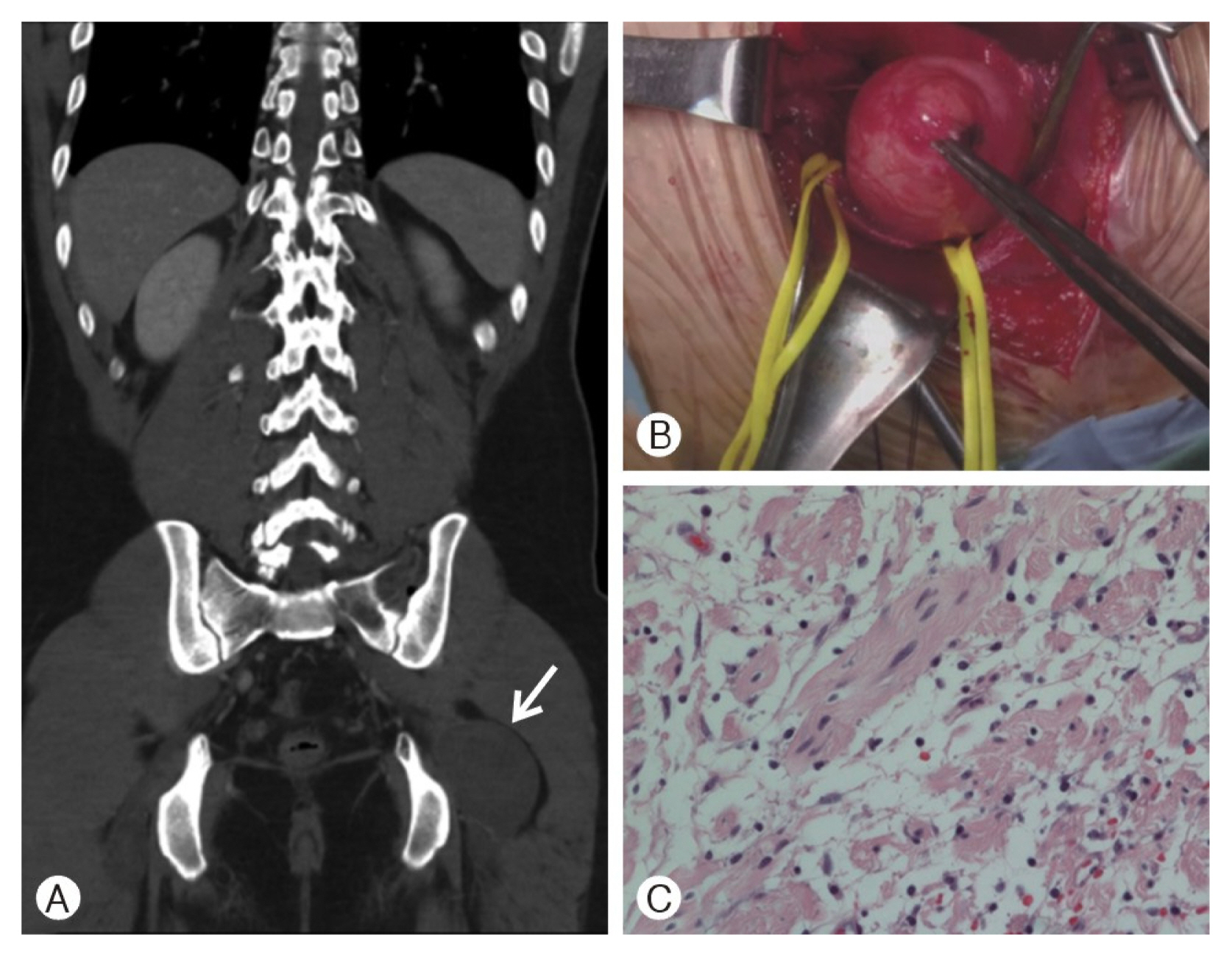
A 23-year-old man with NF1 diagnosed 6 years prior presented with left hip pain. Two round-shaped tumors were found on abdominal computed tomography (CT) and magnetic resonance imaging (MRI). Tumor sizes were 4.2×3.3 cm and 7.0×4.9 cm respectively. To reduce left hip pain, we decided to remove these tumors. The patient underwent two separate excisional operations. One originated from the left femoral nerve and the other originated from the left sciatic nerve. Pathologic diagnosis was neurofibroma (Fig. 1). After surgery, hip pain resolved and there were no neurologic complications and no evidence of recurrence on 1-year follow-up CT.
Case 2
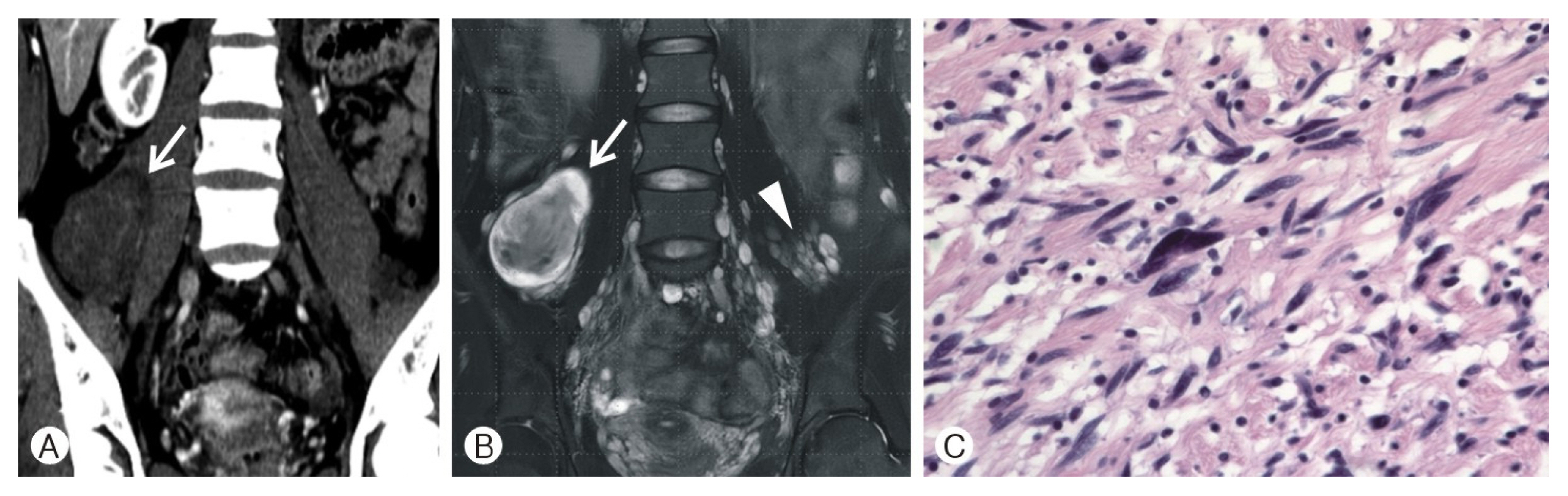
A 25-year-old woman with NF1 diagnosed 7 years prior presented with abdominal pain. On abdominal CT and MRI, a 6.8×4.5-cm-sized and oval-shaped mass was found in the right retroperitoneal space with multiple nodular lesions. The size of this mass and the numerous nodular lesions were suspicious for malignancy. For relief of abdominal pain and pathologic confirmation, we performed surgical resection of the mass. The pathologic findings were moderate cellularity with nuclear atypia, leading to a diagnosis of atypical neurofibroma (Fig. 2). Abdominal pain was relieved postoperative without complications. On 1-year follow-up CT and MRI, there was no evidence of tumor recurrence or malignant changes.
Case 3
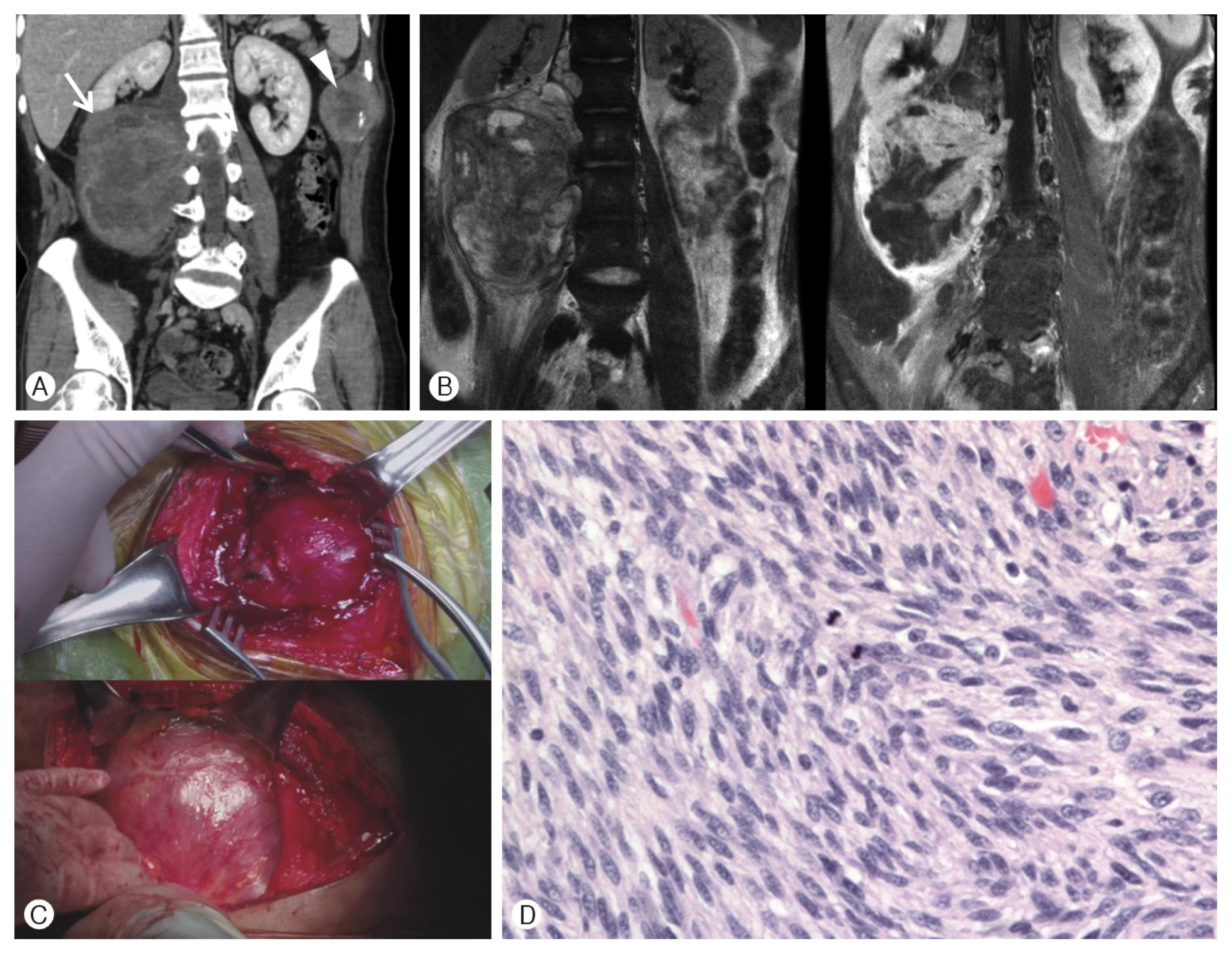
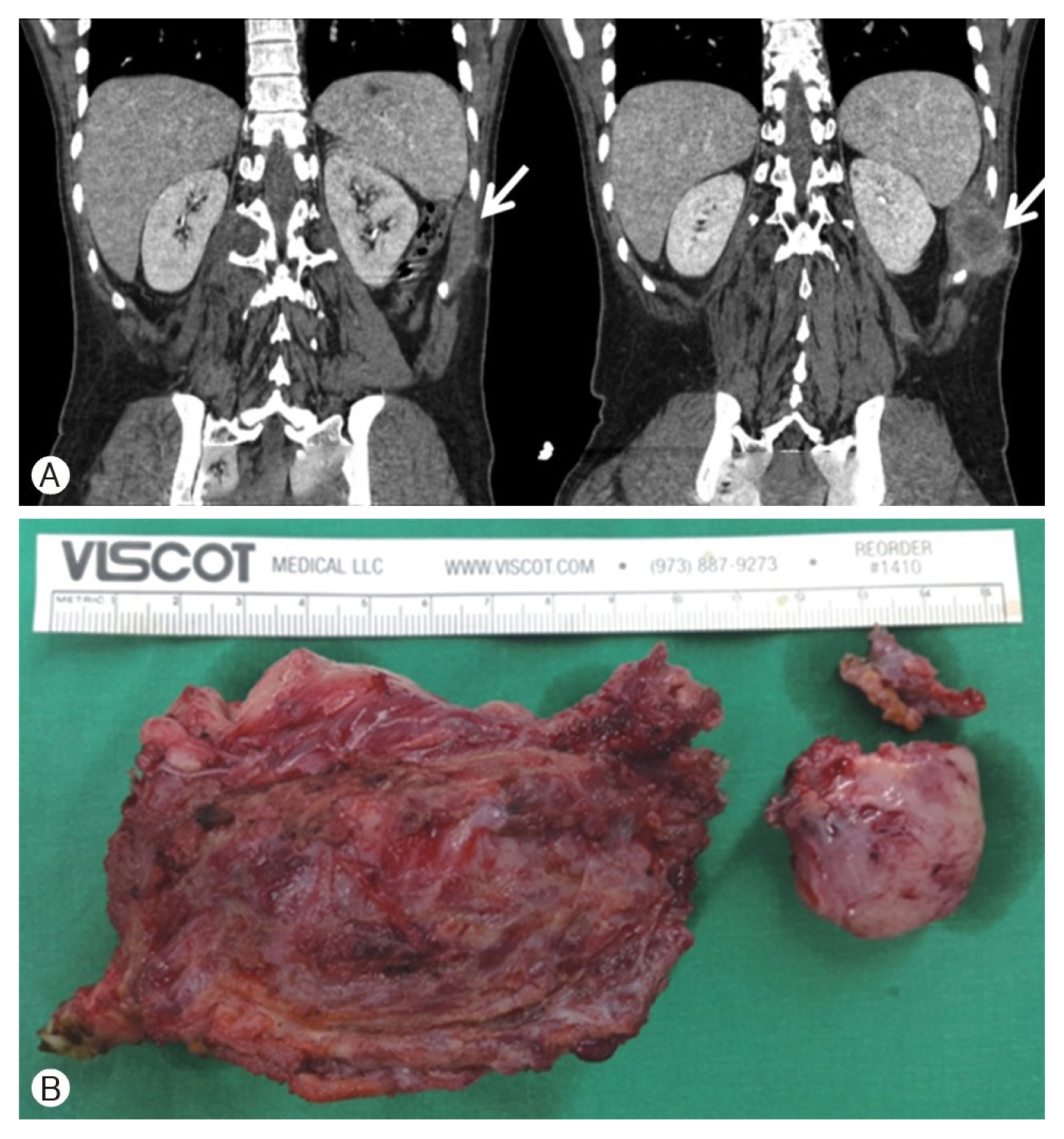
A 22-year-old man with NF1 diagnosed 6 years prior presented with left flank pain. We found 5.1 × 4.1-cm-sized and 12.8 × 9.2-cm-sized tumors on abdominal CT and MRI. Radiologic findings were large-sized tumors with perilesional edematous changes and peripheral enhancement that are suggestive of malignancy. He underwent two separate excisional operations. These tumors originated from the left intercostal nerve and right lumbar plexus and were pathologically diagnosed as MPNST with cellular atypia and numerous mitoses (Fig. 3). The patient received adjuvant radiotherapy, but follow-up CT revealed tumor recurrence. The patient underwent reoperation 2 months after the first operation (Fig. 4). Postoperatively, he received chemotherapy, but 3 months later the patient died from tumor progression and complications.
DISCUSSION
MPNST is a rare disease that is very concerning due to its poor prognosis. The incidence of MPNST is approximately 0.001% in the general population. The recurrence rate is approximately 40% to 65%, and metastasis is found in 16% to 39% of patients. The 5-year survival rate is approximately 25% to 42%2,3,7). Factors related prognosis include tumor size, the extent of resection in surgery, and rate of mitosis. Total resection or wide tumor excision is considered appropriate because chemotherapy and radiotherapy tend to be ineffective2,7). In case 3, we performed surgical resection, radiotherapy, and chemotherapy, but the patient experienced extremely rapid progression of malignant tumor. Despite potential perioperative complications including neurologic deficits, pathologic confirmation and surgical treatment of MPNST is important.
NF1 patients are at high risk of neoplastic disease; the overall risk of cancer in NF1 patients is 2.7 times higher and the risk of malignancy in 50-year-olds is approximately 20%11). Also, approximately 50% of MPNSTs are related to NF1. In NF1 patients, MPNST is less rare than in the general population. The incidence is around 4.6% and the lifetime risk is 8% to 13%. Furthermore, the prognosis of MPNST with NF1 is poorer2,3). Therefore, appropriate surveillance is required for NF1 patients. A particularly close follow-up examination is necessary in cases of growing neurofibroma.
We observed nuclear atypia without mitotic activity on histology in case 2. One-year follow-up CT and MRI revealed no recurrence. However, atypical cell can be found in gray zone between benign and malignant lesions, and transform into malignant tumors in specific genetic characteristics such as deletion of CDKN2A/B. Therefore, atypical neurofibroma is considered a premalignant tumor1,9). Because of the possibility of malignant change, detection and treatment of atypical neurofibroma are important in NF1 patients. Serial MRI and 18F-fluorodeoxyglucose-positron emission tomography can be used for detection of atypical neurofibroma10).
Before pathologic confirmation, imaging findings are used for the differentiation of benign and malignant lesions. Differentiation using image findings is helpful for treatment planning. Previous studies described MRI findings indicative of malignancy, including large size, a peripheral enhancement pattern, perilesional soft-tissue edema, and intratumoral cystic lesions8,12). In case 3, these features were observed on MRI before surgery, leading to suspicion of MPNST. Additional radiotherapy was planned. In case 2, there was no peripheral enhancement pattern, perilesional soft-tissue edema, or intratumoral cystic lesions. However, the large tumor size and distinct multiple nodular lesions on MRI suggested a malignant or atypical lesion6). Our experience suggests that MRI findings are important and useful for the differentiation of three types PNSTs.